
MM-Dist
Software for estimating the distribution of genotypic differences (mismatches) between individuals in a population
Introduction
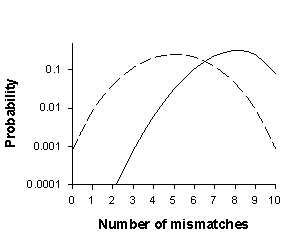
Accurate identification of individuals using molecular markers requires that enough loci are used so that each individual sampled is likely to be unique. When genotyping error is present, it is often worthwhile to use enough loci so that individuals are expected to have multilocus genotypes that differ by more than one locus. MM-DIST is a computer program that calculates probability distributions for how many loci individuals in a population will differ by. For example, the graph below shows the probability of two individuals differing (mismatching) by 0 to 10 loci for unrelated individuals (solid line) and full siblings (dashed line) in a population of bighorn sheep.
References
Please see the following manuscripts for a description of the method:
- Kalinowski ST, M Sawaya, ML Taper (2006) Individual identification and distributions of genotypic differences. Journal of Wildlife Management 70:148-150.
Input files
MM-DIST should read GENEPOP files (either 4 or 6 digit format). There are currently two GENEPOP formats. MM-DIST follows the online format--sample names are read by the first individual in the sample. Alternatively, sample names can be placed after the POP delimiter.
Download
MM-DIST runs on the Microsoft Windows operating system that has the .NET platform installed. See my Software page for instructions on how to install this on your computer (it may already be there).
Click here to download a ZIP file containing MM-DIST and a library of functions (kalinowski_library.dll) that the program needs.
Click here for an example input file (This is the example described in the paper).
Installation / UnInstallation
To "install," place MM-DIST.exe and kalinowski_library.dll in the same folder. Click on MM-DIST.exe to run. Delete both files to "uninstall."